
To me, the idea of living longer is only appealing without the prospect of age-related neurodegeneration. Healthspan is my goal, and the prevention of Alzheimer’s is a major focus.
For decades, research on Alzheimer’s has focused on amyloid-beta plaques and how to reduce them in the brain. Billions have been spent trying to find drugs to combat Alzheimer’s disease by blocking or reducing amyloid-beta (Aβ) plaque formation, but little progress has been made.
A new research model for Alzheimer’s has been published in the journal Alzheimer’s & Dementia. It is an intriguing look at how the research points to Alzheimer’s as an autoimmune disease.
The hypothesis is as follows:
“Alzheimer’s disease as an autoimmune disease” (“AD-squared” or “AD2”). In the AD2 model, AD is explained as a brain-centric disorder of innate immunity involving concurrent autoimmune and autoinflammatory mechanisms. Although AD2 still includes Aβ as an important molecular player, it rejects the “amyloid misfolding hypothesis” per se, instead recognizing Aβ as a physiologically oligomerizing cytokine-like immunopeptide, which is merely one part of a much larger, comprehensive, highly interconnected immunopathic conceptualization of AD.”
Let’s dig into this a bit.
Known risk factors for Alzheimer’s disease include exposure to environmental toxicants (heavy metals, air pollution, and pesticides ), head trauma such as concussions, cardiovascular disease – which causes narrowing of the arteries, smoking, social isolation, depression, and diabetes. Genetics also plays a significant role in this.
Cells can detect foreign invaders and cellular damage in multiple ways:
- Pathogen-associated molecular patterns (PAMPs) are certain types of molecules that cause the immune system to react to a pathogen. For example, specific molecules on the surface of bacteria trigger an immediate immune response, whether live bacteria are present or not.
- Damage-associated molecular patterns (DAMPs) are molecular patterns the immune system recognizes as being given off by damaged cells that need to be killed and cleared out.
Amyloid-beta (Aβ) is synthesized and released in the body as an early responder and reacts to a number of PAMPs and DAMPs. Amyloid beta triggers the innate immune system cascade.
Microglia are immune cells found in the brain and spinal cord. They play a critical role in maintaining the health and function of the central nervous system. Microglia are the first line of defense against invading pathogens and harmful substances in the brain.
Amyloid-beta augments microglial activation and pro-inflammatory cytokine release. It does this through interacting with NLRP3 (inflammasome activator) and TREM2.
The paper hypothesizing Alzheimer’s as an autoimmune disease explains that amyloid-beta attacks normal neurons due to “electrophysiological similarities between neurons and bacteria in terms of transmembrane potential gradients (–80 mV) and anionic charges on outer leaflet membrane macromolecules (gangliosides in neurons; cardiolipin or lipopolysaccharides [LPS] in bacteria), which renders them similarly susceptible to necrosis from cytotoxic membrane-penetration by antimicrobial peptides (AMPs) such as Aβ.”
That’s a lot to grasp. If I understand correctly, amyloid-beta deposition in the neurons is due to the false detection of neurons as LPS (also known as endotoxin) or other invaders, which triggers a PAMP response to bacteria. The breakdown of the neuron produces DAMPs, which then trigger more amyloid-beta to adjacent neurons.
Going one step further:
“This chronic self-sustaining innate autoimmune attack upon self-neurons (during which Aβ cannot distinguish between host and foreign cells) results in pathological prolongation of Aβ’s dual immunomodulatory and antimicrobial actions, perpetuating ongoing immunotoxicity and neurotoxicity particularly at the functional level of the synapse, which is clinically relevant given the importance of synaptic transmission to information processing and the observation that disordered information processing is central to the symptoms that characterize AD’s clinical presentation (i.e., altered memory, cognition, executive functioning, judgment). Because glial cells are anatomically associated with synapses, and control synaptic formation, function, plasticity, and elimination, the effects of Aβ upon TREM2, NLRP3, and GAG immunomodulatory receptors are additional key elements of Aβ-mediated synaptotoxicity within the AD2 model.4 Chronic pro-inflammatory cytokine release, tau aggregation, and mitochondriopathy with oxidative stress further contribute to disease progression in the AD2 model.”
Why is this such a unique theory?
Autoimmune diseases, for the most part, are due to the adaptive immune system mistaking a particular type of cell as foreign. In celiac disease, for example, exposure to gluten leads to the production of various antibodies and the activation of immune cells like T cells. T cells, in turn, release inflammatory cytokines that contribute to intestinal tissue damage. The adaptive immune response is highly specific to remembering what is ‘foreign’ and attacking it when it sees the foreign protein again.
In this case of Alzheimer’s as an autoimmune disease, the researchers look at the innate immune system as attacking ‘self’ neurons. However, we aren’t talking about autoantibodies here. Instead, the innate immune response is usually an immediate response to cell injury, and usually unintended innate immune responses are called autoinflammatory diseases (e.g., familial Mediterranean fever).
The researchers state that Alzheimer’s disease is not an autoinflammatory disease, though. “However, as conjectured in AD2, AD is truly an autoimmune disease of innate immunity because Aβ, released as an antibacterial immunopeptide, inadvertently (but specifically) mistakes a neuron as non-self, puncturing its membrane as it would a bacterium.”
Where does this leave us?
This theory proposes new avenues of research. Specifically, the researchers are calling for more research on the roles of l-tryptophan and l-arginine metabolism.
“In the AD2 mechanistic model, AD is an autoimmune and autoinflammatory disorder of innate immunity, subject to regulatory control by amino acid (L-tryptophan and L-arginine) metabolic pathways.”
The researchers give this overview of how the parts come together:
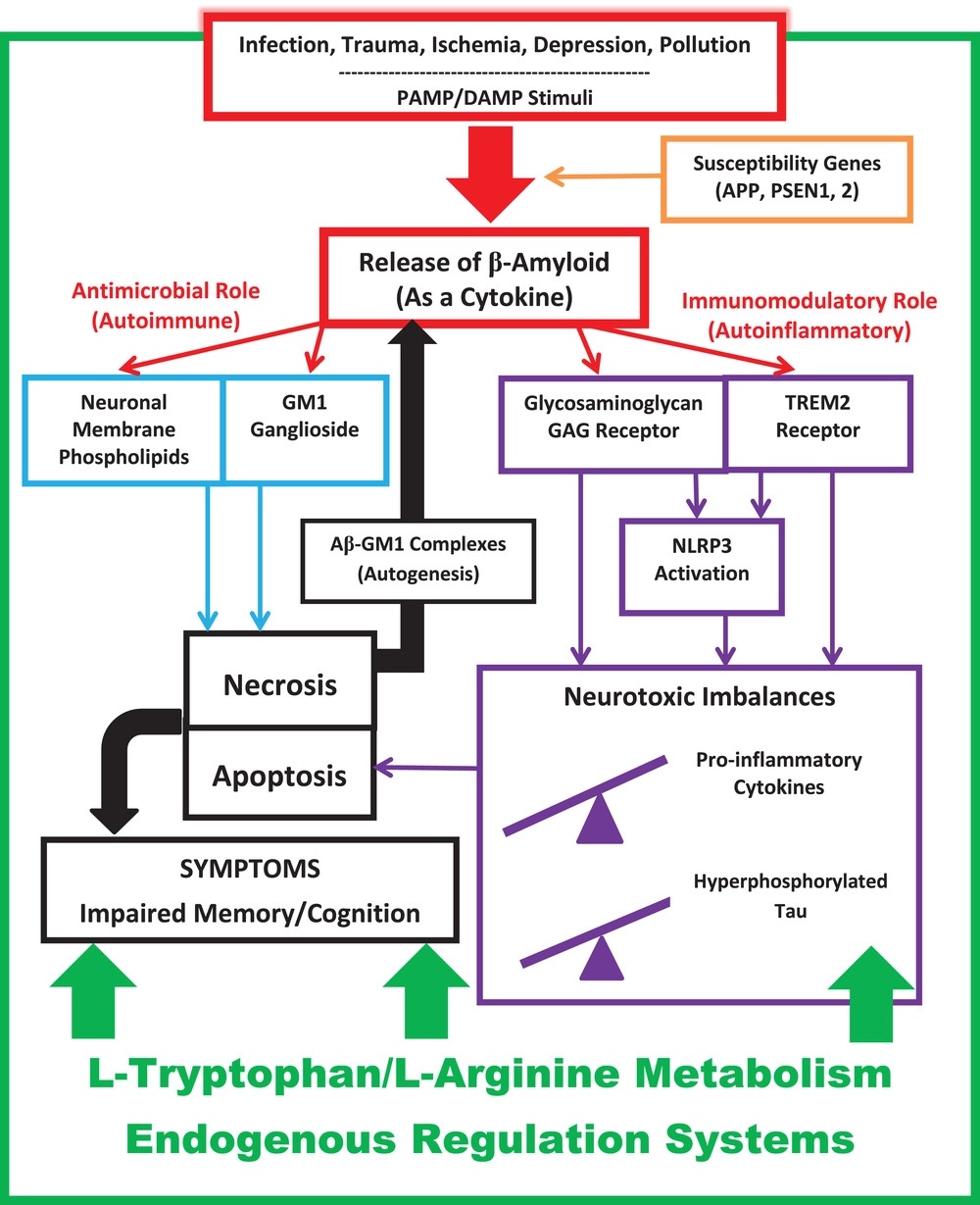
I encourage everyone interested in longevity and cognitive function to read through the full article: https://alz-journals.onlinelibrary.wiley.com/doi/10.1002/alz.12789
This ties together with other recent research on an Alzheimer’s trial with four common supplements – L-serine nicotinamide riboside, N-acetyl-L-cysteine, and L-carnitine tartrate. For example, l-serine (an amino acid) interacts with l-arginine in the formation of nitric oxide.